⇪
| 09/10/15 | gitter v1.1.1 released. This includes bug fixes, performance enhancements and general improvements. |
To begin using gitter make sure you're using R version >3.0. If you do not have R, you can download it from here as well as RStudio, which is a useful user interface for R
Once you have R/RStudio up and running, make sure you have the EBImage package is installed from Bioconductor like this:
source("http://bioconductor.org/biocLite.R")
biocLite("EBImage")
then install and load gitter:
install.packages('gitter')
require(gitter)# Run a simple demo
gitter.demo(1);
# Run a simple demo
gitter.demo(2);
To get started, let's process a single image

# Get the sample image path
f = system.file("extdata", "sample.jpg", package="gitter")
# For your own images, just supply the path to the image:
# f = "/path/to/my/file.jpg"
# Process it, giving information on the progress
dat = gitter(f, plate.format=1536, verbose='p')
# View head of the results
head(dat)
It's that simple! Two output files are saved to your working directory.
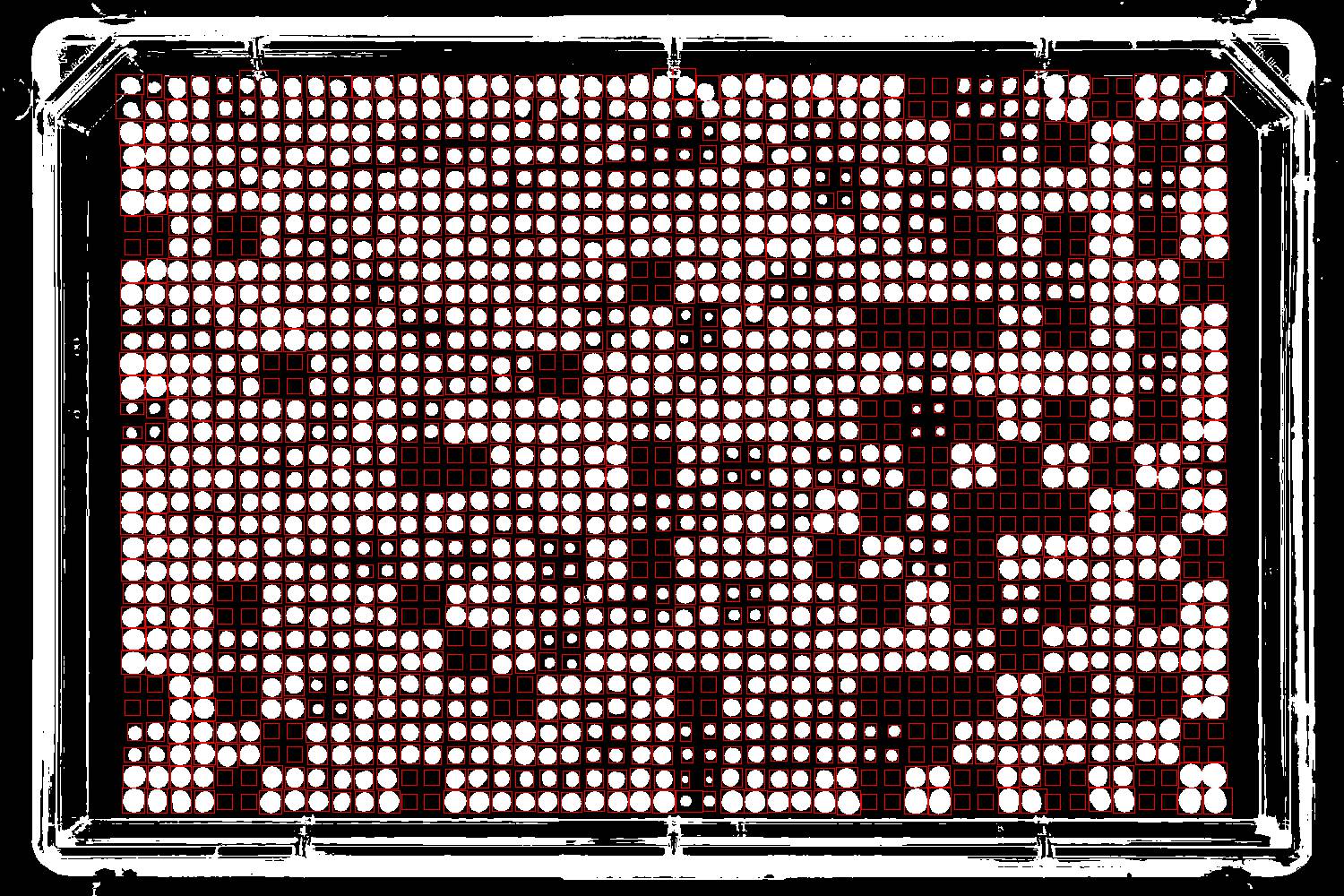
You may sometimes have plates with colonies growing very sparsely. Such images are usually difficult to process, since they often have completely empty rows or columns. Here is an example:

gitter has a feature which will let you process these sparse images. To do so, you'll need an image of a plate with with more dense colonies taken with the same dinensions and positioning. We call this the "Reference image".
If gitter can process your reference image, it knows where the colonies are located and can find the corresponding location on the sparse colony plate. To process a sparse colony plate using a reference image use the gitter.batch command. Here is an example:
# Get the reference image path
r = system.file("extdata", "sample.jpg", package="gitter")
# Get the sparse plate image
s = system.file("extdata", "sample_dead.jpg", package="gitter")
# Process it, giving information on the progress
gitter.batch(image.files=s, ref.image.file=r,
plate.format=1536, verbose='p')
Or you can try out a quick demo:
gitter.demo(2)Here is a visual representation of how this works:

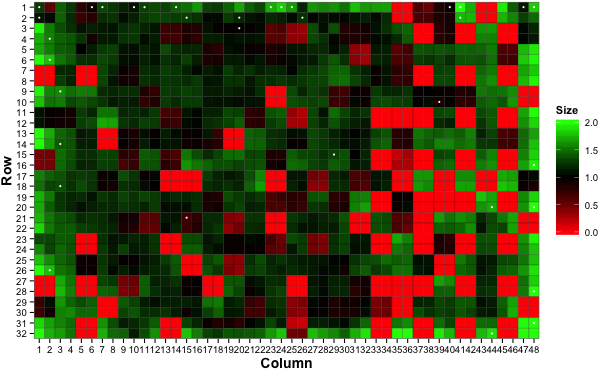
For visual validation, gitter has a built in plotting function which allows you to generate a heatmap and/or a bubble plot
# Plot a heatmap
gitter.plot(dat, type='heatmap')
# Plot a bubble plot with small colonies as black
# and large colonies as red
gitter.plot(dat, type='bubble', low='black', high='red')
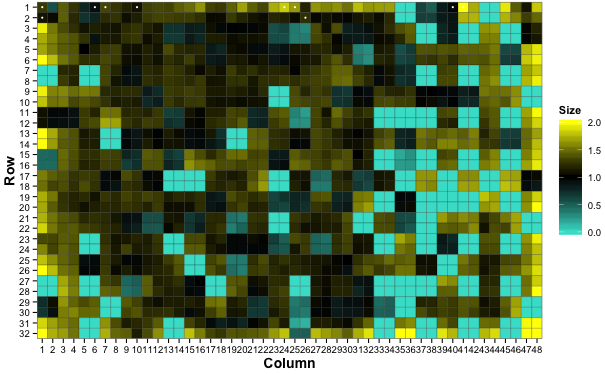
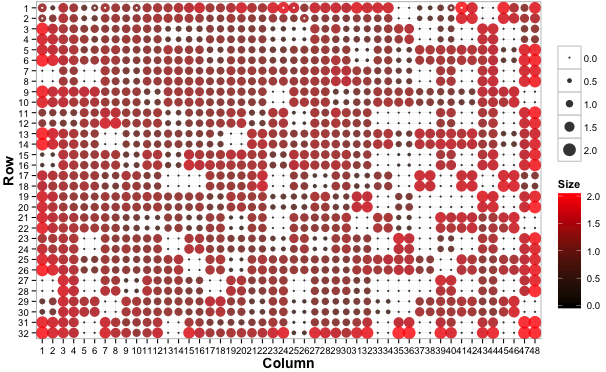
White dots on the plots represent colonies flagged to be dubious (see documentation).
You can adjust the colours defining the colony sizes as needed
# Plot a heatmap with custom colours
gitter.plot(dat, type='heatmap',
low='red', mid='black', high='green')
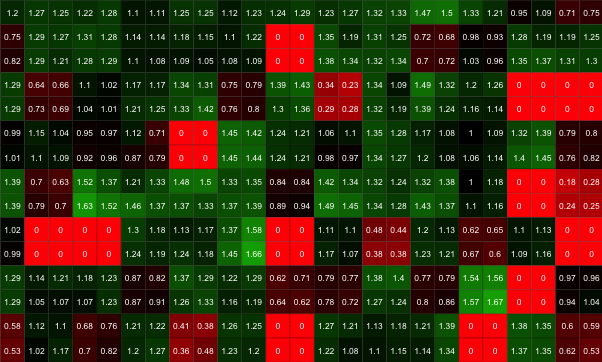
and overlay colony size values
# Plot a heatmap with custom colours and overlaid size values
gitter.plot(dat, type='heatmap',
low='red', mid='black', high='green', show.text=T)
For further documentation on the parameters or how to process batch images view the pdf manual on CRAN or type the following into your R console
?gitter
?gitter.batch
?gitter.read
?plot.gitter
For information on the package
?Rgittergitter was developed by Omar Wagih and Leopold Parts at the Donnelly Centre for Cellular and Biomolecular Research, University of Toronto
A lot of time and effort went into developing gitter. If you use it in your research, please cite:
Wagih, O. and Parts L. (2014) gitter: a robust and accurate method for quantification of colony sizes from plate images. G3 (Bethesda) pii: g3.113.009431v1
PUBMED
If you have a suggestion, or feature request please post it in the issues section. If you're having trouble with gitter, check out the documentation as mentioned above or contact us and we’ll help you sort it out